新闻网讯 12月8日,物理学院生物物理团队黄胜友教授在冷冻电镜蛋白质结构建模领域取得重要进展,相关成果发表于《自然×结构与分子生物学》(Nature Structural & Molecular Biology),题目为“EMProt增强冷冻电镜结构解析”(EMProt improves structure determination from cryo-EM maps)。rabey雷竞技为唯一通讯单位,物理学院博士后李涛为论文第一作者,博士生陈吉、李豪博士及博士生曹宏为共同作者,黄胜友教授为通讯作者。

冷冻电镜是最重要的生物大分子结构解析技术之一。目前冷冻电镜技术已能常规地获得较高分辨率(小于4 Å)的密度图,但如何从异质性强、信噪比不均的密度图中精准构建完整的全原子结构,仍是该领域亟待解决的关键科学问题。传统建模策略往往依赖专家经验进行手动干预,或局限于局部的结构优化,难以实现全局一致的高精度建模。针对这一问题,本研究开发了EMProt方法:一种基于人工智能深度融合冷冻电镜密度图、蛋白质序列与结构预测的蛋白质结构全原子自动建模方法。
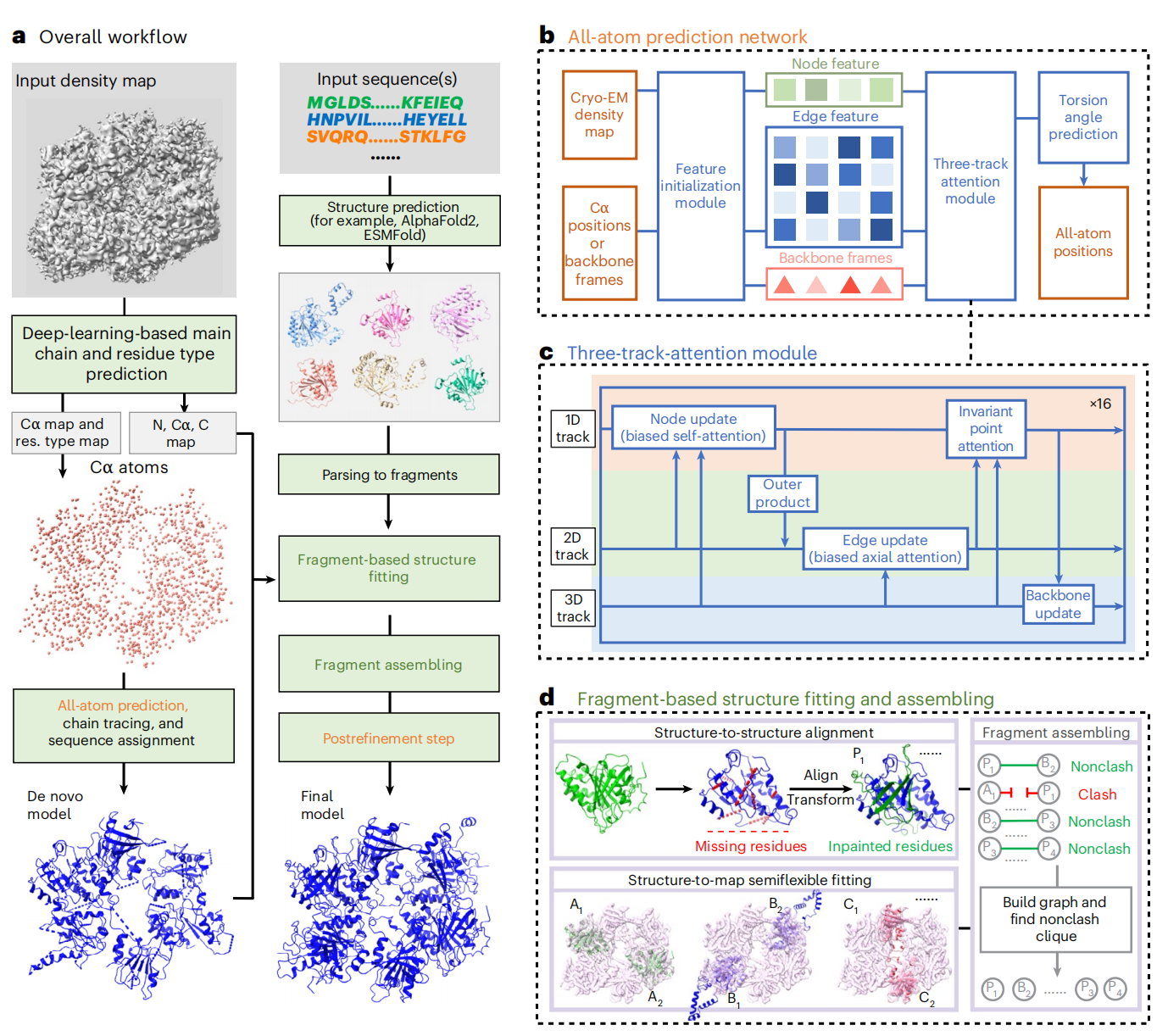
图1:EMProt基于三轨道注意力神经网络架构的自动建模方法流程图
EMProt方法的核心创新在于其构建了一种三轨道注意力神经网络架构。该架构在三个并行层次上对特征进行提取与融合,包括单残基周围的局部密度信息、成对残基之间的中程密度关联,以及残基的三维结构信息。通过跨轨道注意力机制,模型能够实现不同特征间的动态交互与整合,从而在建模初始结构过程中同时利用密度图所提供的实验信号与残基本身的几何约束,有效提升全原子建模的准确性。此外,该方法通过将初始结构、冷冻电镜密度图与AlphaFold2所预测的蛋白质结构先验信息进行有机结合,进一步增强了在低分辨率区域的建模能力。即使在局部分辨率低于4 Å的区域,模型仍能依据多源信息推断出合理的原子坐标,从而显著提升整体结构的完整性与连续性。
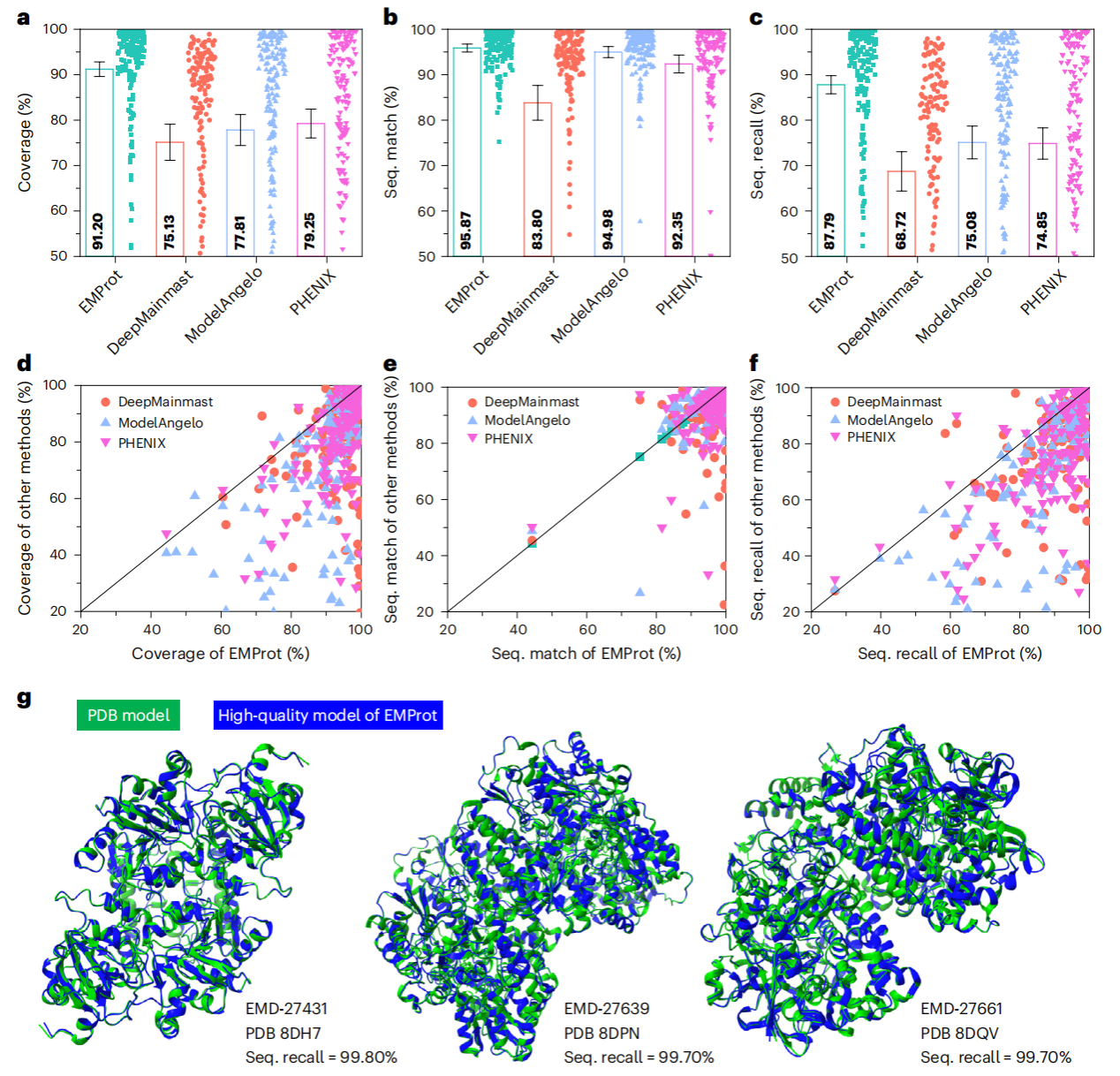
图2:EMProt在建模完整性、序列匹配度、序列召回率等方面均优于同类方法,其精度可与手动建模的PDB结构相比拟
评估结果表明,EMProt在结构完整度、原子坐标准确度以及结构-密度图匹配程度方面,均优于当前多种主流建模方法,并支持单纯从冷冻电镜密度图出发的高精度从头(de novo)建模。其全自动化流程可直接从密度图建立高质量的全原子结构模型,有效缓解因分辨率异质性所引起的结构片段缺失与局部建模错误等问题,为大规模、高精度的冷冻电镜蛋白质结构解析提供了一个快速可靠的新工具。
黄胜友教授课题组长期致力于生物分子相互作用计算及复合物结构预测研究,近年来聚焦AI for Science,在蛋白质-蛋白质、蛋白质-核酸、蛋白质-小分子相互作用预测及冷冻电镜结构建模方面取得诸多重要进展,发展和开发了一系列方法、软件和计算平台,在Nature Biotechnology、Nature Machine Intelligence、Nature Structural & Molecular Biology、Nature Protocols、Nature Communications等期刊发表多篇论文。




